4 ECD Stability Under Activating Conditions
Despite the fact that the TMD remains elusive, my hope was that investigating conditions thought to open the channel I might observe large-scale ECD rearrangements like those observed in ASIC. In this chapter I cover my efforts on that front, starting with a map of mouse ENaC solved pre- and post-trypsin treatment. Next, I investigate a putative sodium self-inhibition site that had been described previously in the literature (including by our own lab) in the presence of Na+ and K+. Finally, I highlight the extensive newly-resolvable surface glycosylation present in all of my ECD maps.
4.1 ECD conformation is unchanged by trypsin cleavage
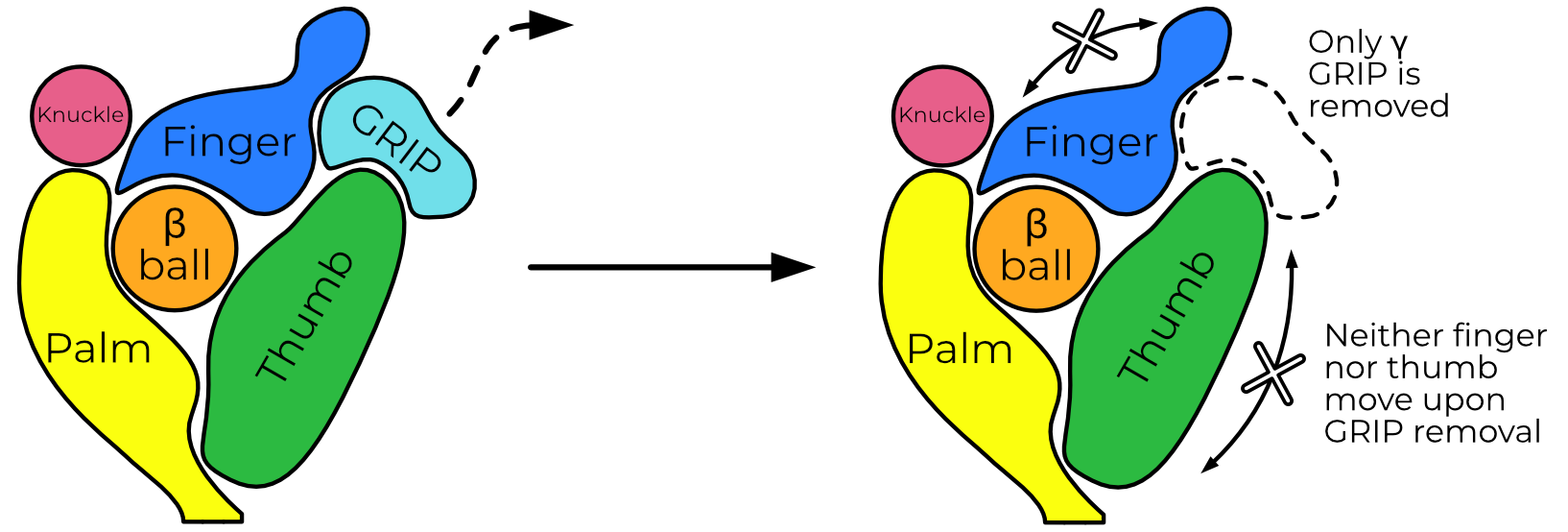
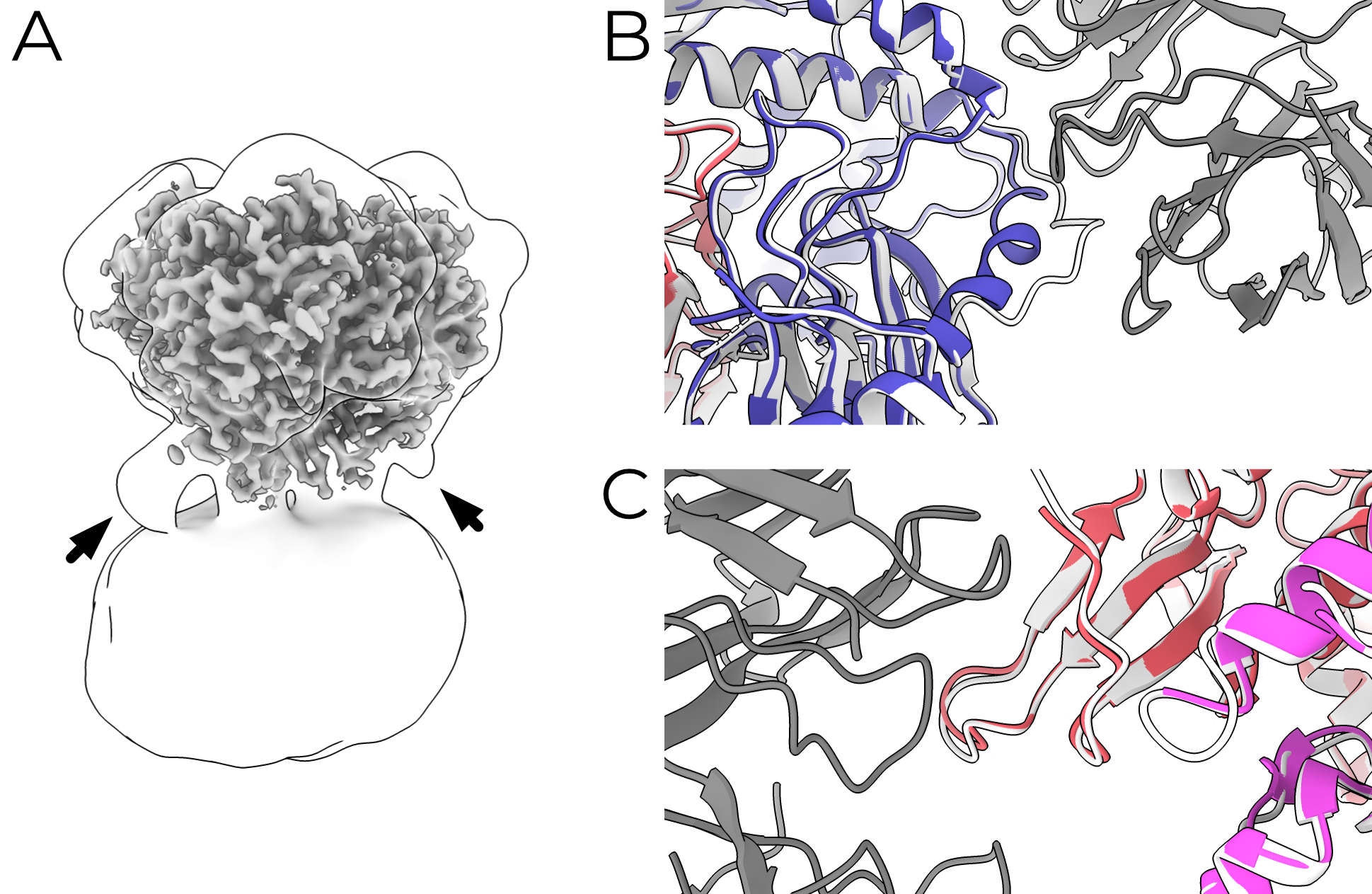
ENaC can reliably be opened by trypsin treatment1. Additionally, the inhibitory peptide is easily resolved in all of my ECD maps. I wondered whether treatment with trypsin would result in movement of the finger and thumb domains as the model of ASIC-like gating movements predicts (Figure 4.1). It’s important to note here that I had significant help from Arpita Bharadwaj, who performed the purification and initial characterization of the mouse material for these grids. Both uncleaved and trypsin-treated mouse ENaC were of similar map quality (3.24 and 3.30 Å respectively with whole-protein masks).
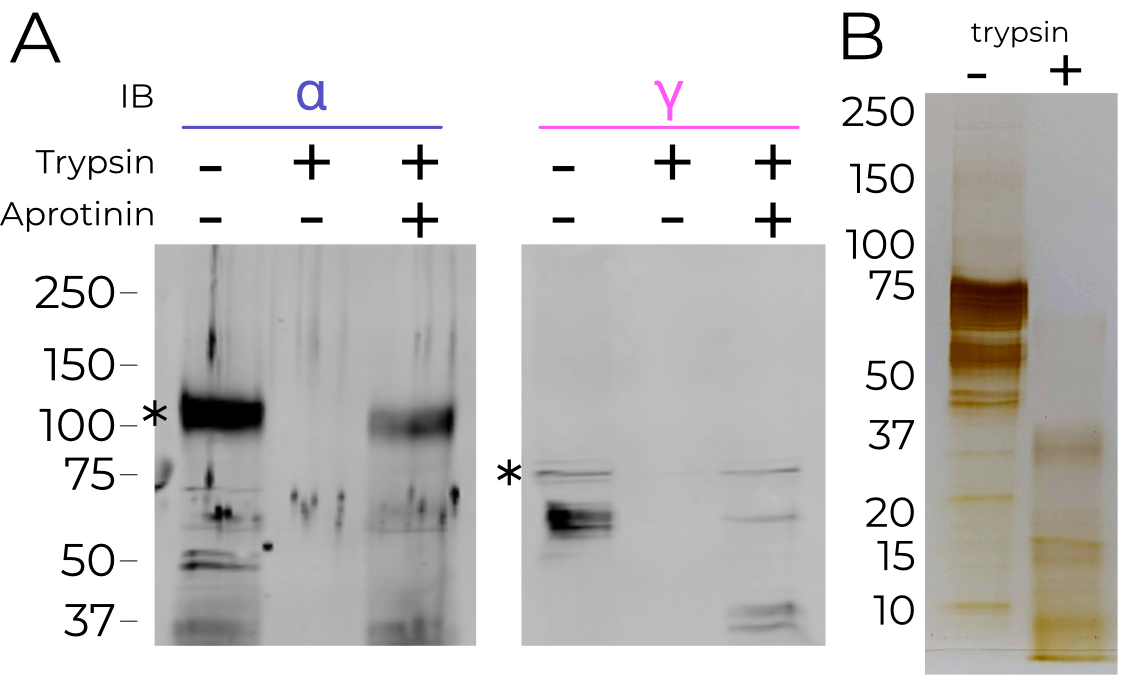


Biochemical analyses show that trypsin treatment cleaves both the α and γ subunits (Figure 4.2). Additionally, trypsin treatment of X. laevis oocytes expressing mouse αβγENaC show the expected increase in amiloride sensitive current (Figure 4.3). However, aside from the disappearance of the inhibitory peptide, the ECD is otherwise superimposable (RMSD 0.440 Å; Figure 4.4). The finger and thumb do not shift even a fraction of what the ASIC-like model would predict.
Based on the observed cryoEM map density, only the γ subunit GRIP domain is released from the channel. However, biochemical characterization indicates both are digested by trypsin. I also note that the entire GRIP domain, not just the inhibitory peptide, has reduced map density in the trypsin-treated sample. This suggests that the entire grip domain might be released from the subunit, but of course it’s equally possible that release of the inhibitory peptide results in increased GRIP flexibility, which blurs out the map.
These results are incompatible with a large-scale movement of the ECD upon trypsin digestion. The existence of some γ GRIP density indicates that the trypsin-treated map is an ensemble of particles which have and have not released the γ GRIP domain. However, I do not observe any blurring of the γ thumb, which would be expected in a map comprising particles with significantly shifted conformations. I must therefore conclude that the removal of the γ inhibitory peptide is not sufficient, on its own, to induce such changes. Additionally, I note that these maps are the first indication that trypsin processing of the α subunit does not release its GRIP domain. This is surprising but perhaps not wholly unexpected; the γ subunit is known to be essential for activation by trypsin but the same has not yet been shown for αENaC3.
Biochemical analysis indicates that ENaC is highly cleaved by trypsin, degrading the channel to fragments 37 kDa and smaller. It may be that the material in our map is highly cleaved, but held together by surface contacts throughout the channel which are disrupted by the addition of SDS from the loading buffer. This hypothesis could be tested with a native gel.
4.1.1 A strained tyrosine in the thumb
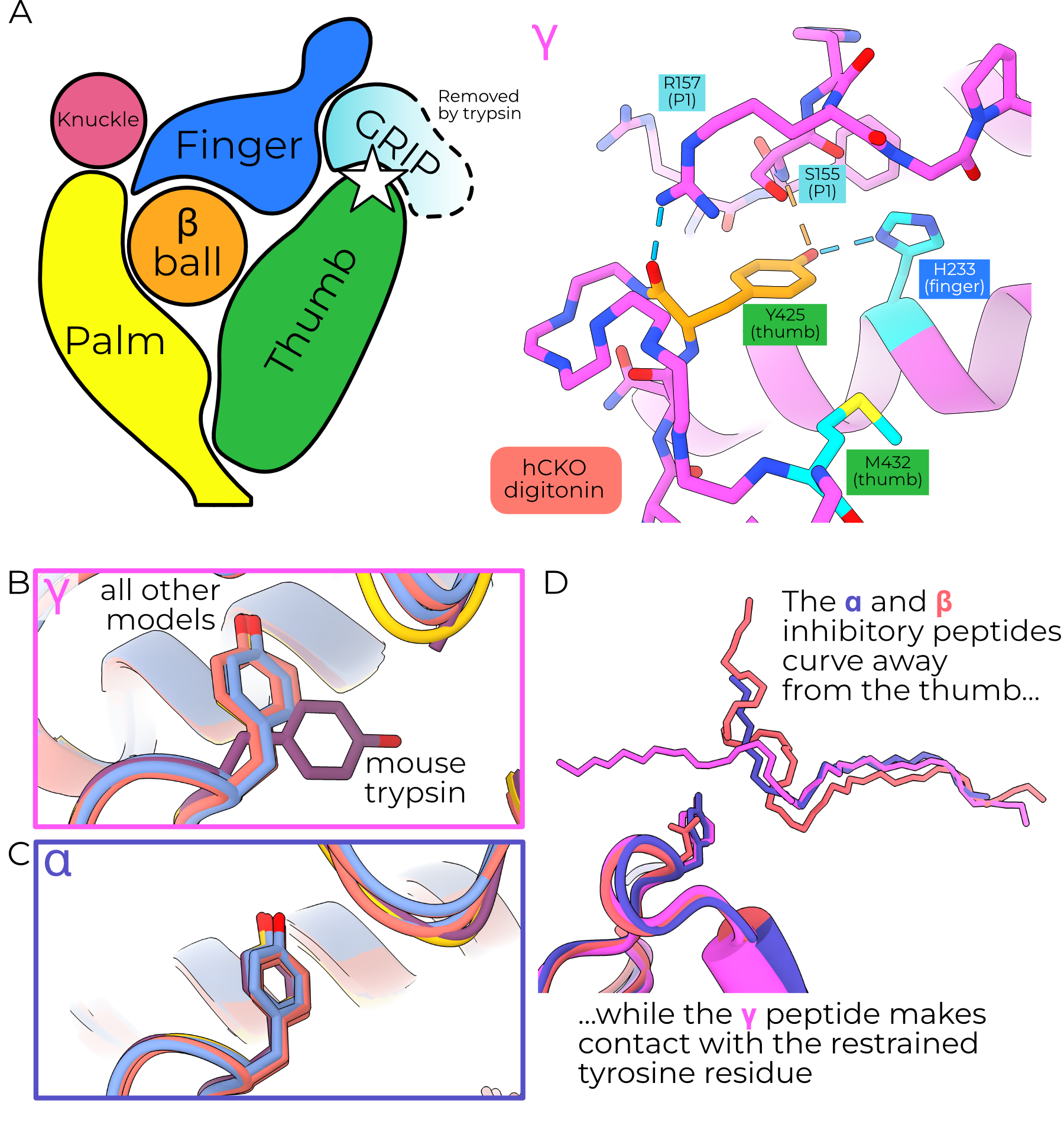
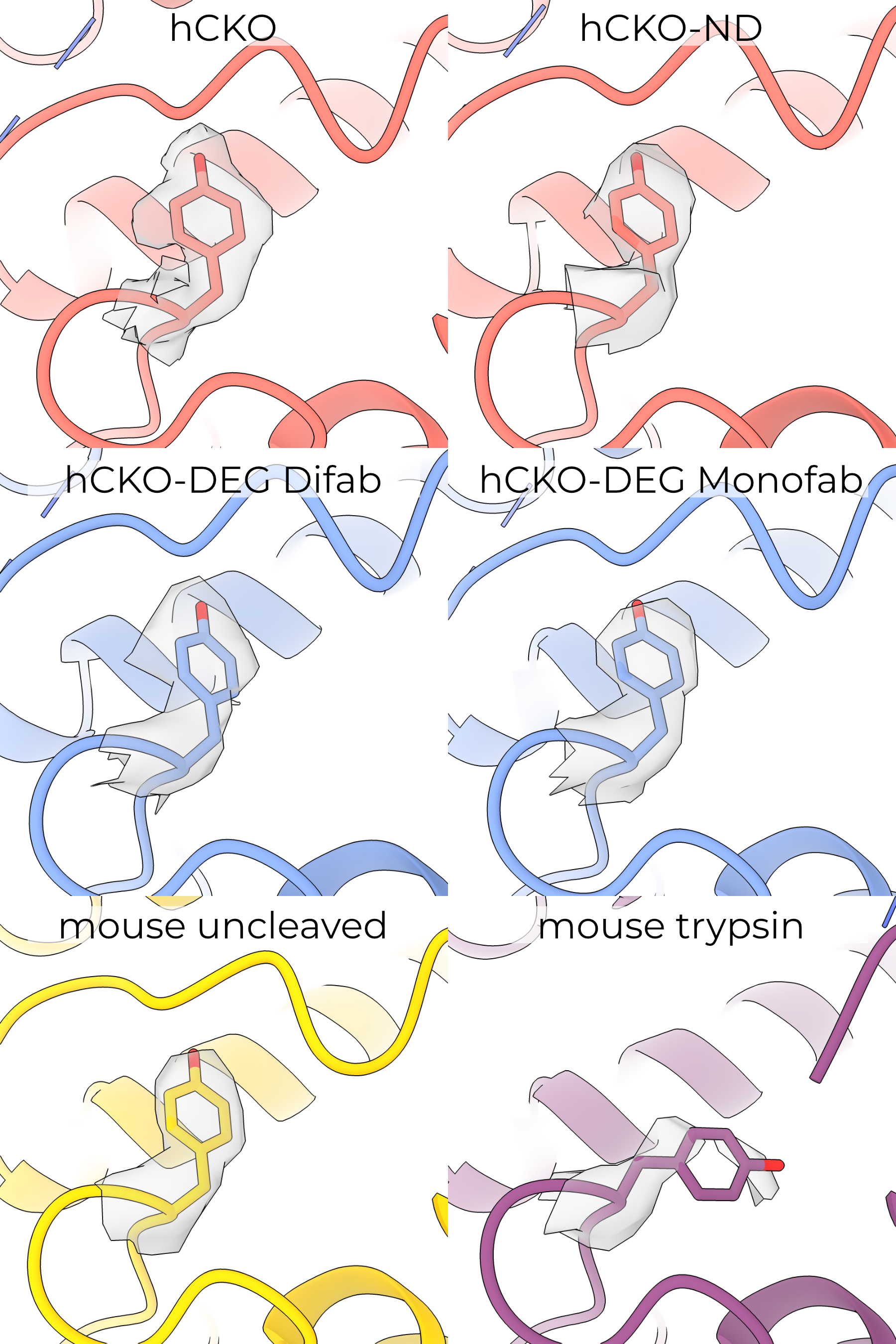
The high-resolution maps of the γ ECD reveal an interesting rotamer outlier, γY425 (Figure 4.5). The map for this residue is of high quality in five of the six maps, supporting its unlikely (prior probability of 0.035%) conformation (Figure 4.6). The single map with poor quality is the trypsin-treated mouse map, in which the density is blurred and unclear. Human γY425 forms hydrogen bonds with the finger (H233) and inhibitory peptide (Figure 4.5 A). This tyrosine adopts the same pose in all models except the trypsin-treated mouse model, in which it may rotate to point toward the outer GRIP domain (Figure 4.5 B). Given the poor quality of γY431 (the equivalent position to human Y425) in the mouse trypsin map, I cannot assign this rotamer with certainty.
The equivalent residue in α is also a tyrosine (αY447 and 474 in human and mouse respectively), but the inhibitory peptide curves away from the channel center, removing the interactions with the backbone and aromatic oxygen atoms and allowing a more favored rotamer (Figure 4.5 C and D). The same position in β, N417, only forms a hydrogen bond with the finger domain (N225). The lack of any interactions between βN417 and the βENaC GRIP domain, which is not cleaved to open the channel, is further suggestive of this residue’s role in proteolytic gating.
I note also that the map quality worsens for the nearby γM438, mutation of which alters channel gating4. The reduced quality for this map region upon trypsin treatment may represent increased flexibility, perhaps indicating that conformational strain in the upper thumb is released along with the inhibitory peptide.
4.2 Sodium self-inhibition does not rearrange the ECD
I next investigated differences in ECD conformation among protein purified in external K+ instead of Na+. High extracellular K+ does not prime inhibition in wild-type channels and SSI is rapidly attenuated upon exposure to low (i.e., <10 mM) Na+4,5. Protein purified in K+ buffer with nominally zero Na+ should therefore be completely uninhibited.
The linker connecting β6 and β7 is important for SSI, and our lab has previously reported evidence of a putative bound Na+ ion in this region5,6. Moreover, cross-linking of several pairs of amino acids in the ECD affects SSI magnitude, implying that some rearrangement is required for SSI5,7. Given this body of research, I solved structures of ENaC in high (200 mM) and low (nominal 0 mM Na+, 200 mM K+) sodium. I expected the high-Na+ model to show rearrangements throughout the ECD, as would be required to transfer Na+-binding state to the pore.
Our maps of human CKO ENaC in Na+ and K+ buffers achieved resolutions of 2.3 and 3.0 Å, respectively. The Na+ structure was solved in digitonin with both the α- and β-binding Fabs, while the K+ structure was solved in nanodiscs without any Fabs. Although previously it has proven difficult to break the pseudo-threefold symmetry without including Fabs, I worried that their inclusion may affect SSI. Fortunately, the high quality of the cryoEM data allowed us to break the symmetry with neither Fabs nor an external reference — I believe the major symmetry breaking features are micelle-proximal glycans (Figure 4.7). Binding of either Fab induces minimal rearrangement of ENaC; the β epitope does not change conformation upon binding, while the α epitope pulls the loop from αP264-T274 approximately 4.5 Å away from the protein core without distorting the structure outside that loop.
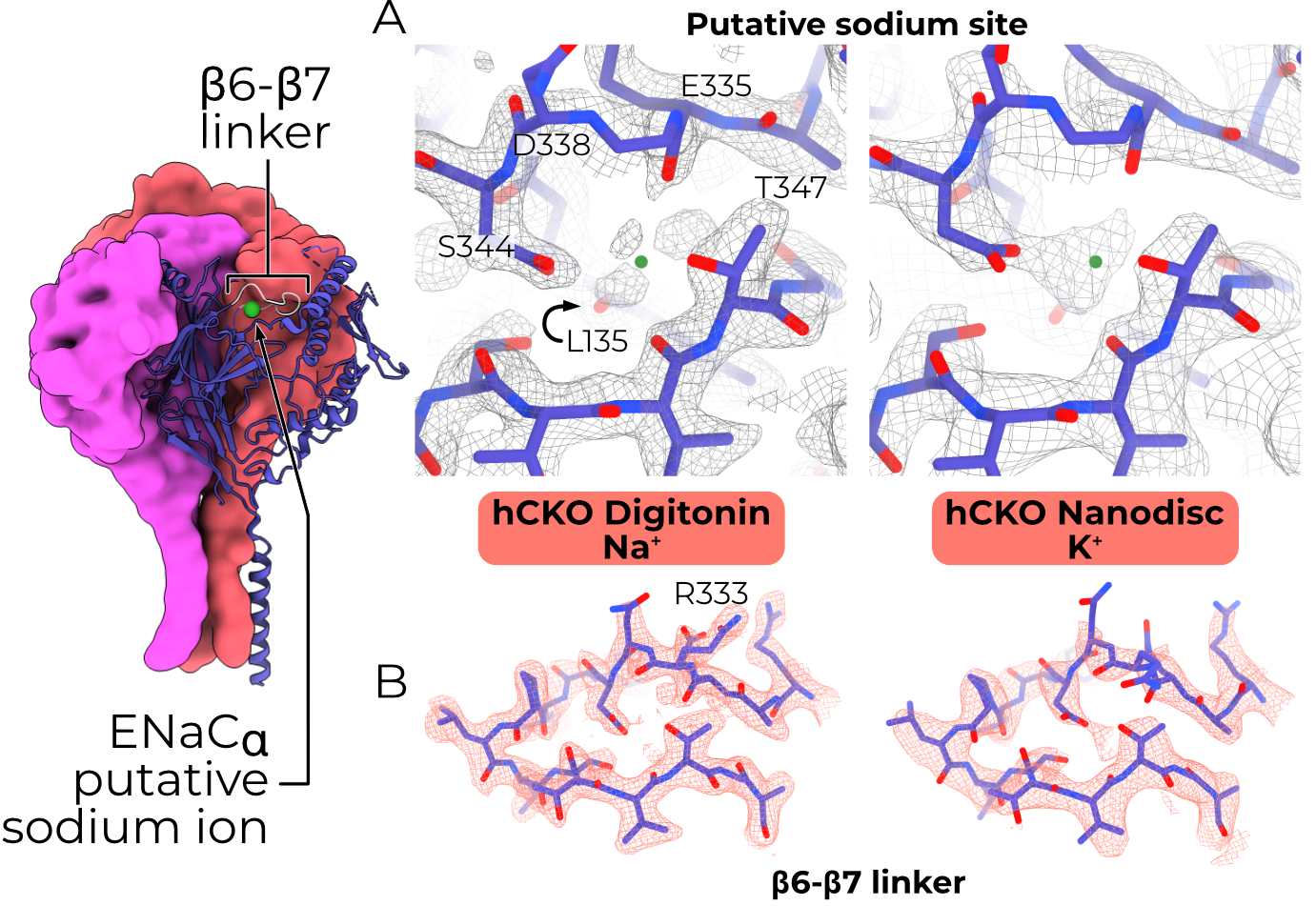
To my surprise, no resolved region of the ECD undergoes any significant structural rearrangement between high- and low-Na+ buffers (RMSD 0.490 Å, Figure 4.8 A). Aside from the changes induced by the α-binding Fab, the only Cα shifts greater than 2 Å are residues terminating the resolved regions of the channel, which are likely a result of poor map quality. An acidic cleft in the αENaC finger, near the β-ball, is implicated in sodium self-inhibition5. However, the cryoEM maps have density for a ligand in the acidic cleft in both the Na+ and K+ maps (Figure 4.8 A), although the site is slightly expanded in the K+ map, perhaps to accommodate the larger K+ ion. The β6-β7 linker has high quality density in both maps, supporting an almost-identical (Cα RMSD 1.076 Å when the whole α subunit is aligned by the palm domain) conformation, aside from the blurring of αR333 across multiple rotamers (Figure 4.8 B).
Although SSI is sensitive to pH, the buffer conditions for the CKO map should be at least 55% inhibited8. Attempts at detecting other classes of particle in the CKO ENaC (Na+) map were not successful, leading us to believe that the particles are homogeneous. The lack of any significant structural difference in the acidic pocket in the presence of Na+ and K+ is more compatible with an SSI model in which Na+-bound ENaC is “locked” in a closed state, rather than some third inhibited state.
4.3 Glycans make important contacts among subunits and with the membrane
Glycosylation of ENaC at conserved asparagine sites is essential for sodium self-inhibition, trypsin activation, and mechanosensitivity9,10. The high-quality maps obtained in this study enable modeling of significantly more and longer glycans than in previous work. I find a total of 19 distinct glycosylation sites, with up to nine modellable sugars in a single glycan.
For the most part, the sugars are identical in all six models and make no protein or glycan interactions. These sugars likely play a role in other regulatory roles, such as channel trafficking and maturation or interaction with the extracellular matrix9–11. However, two classes of glycan are particularly interesting: the micelle-proximal and the intersubunit glycans.
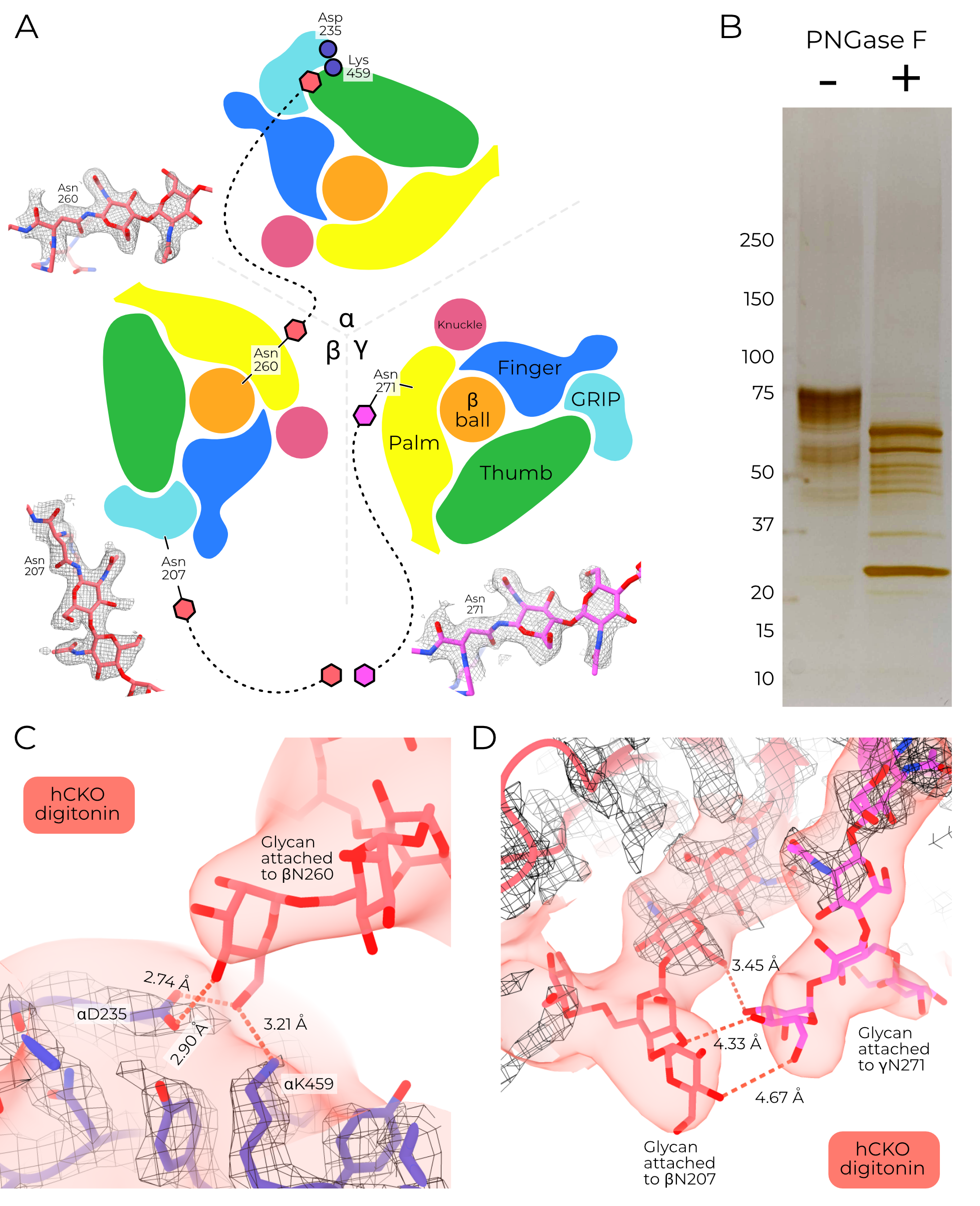
The intersubunit glycans stretch from one subunit to another (Figure 4.9 A). I am able to model long chains for these glycans, with some maps supporting up to nine monosaccharides. Due to the flexibility of these long chains, the terminal side is quite blurred out in the cryoEM maps, and so I cannot be confident about the exact orientation or identity of the constituent sugars. Nonetheless, the maps are of sufficient quality to at least suggest sugar-mediated interactions between subunits, and the first sugars are of sufficient resolution to assign the asparagine residues to which they are coupled.
Treatment with PNGase F induces a size shift in the purified protein, confirming that the protein is glycosylated (Figure 4.9 B). I built typical high-mannose glycans (Man5GlcNAc2) into the models as expected for GnTI- and Sf9 cells to assess these interactions at a low resolution12,13. One glycan reaches from βN260 to the α subunit (Figure 4.9 C), potentially hydrogen bonding with αD235 and αK459. These three residues are absolutely conserved across ENaC genes surveyed. Similarly, glycans attached to conserved sites βN207 and γN271 connect the β GRIP to the γ palm (Figure 4.9 D). Knock-out of sugars in similar positions in αENaC increase blood pressure in mice10. The relatively high quality of these glycans suggest they are quite conformationally stable and may play a role in channel function.
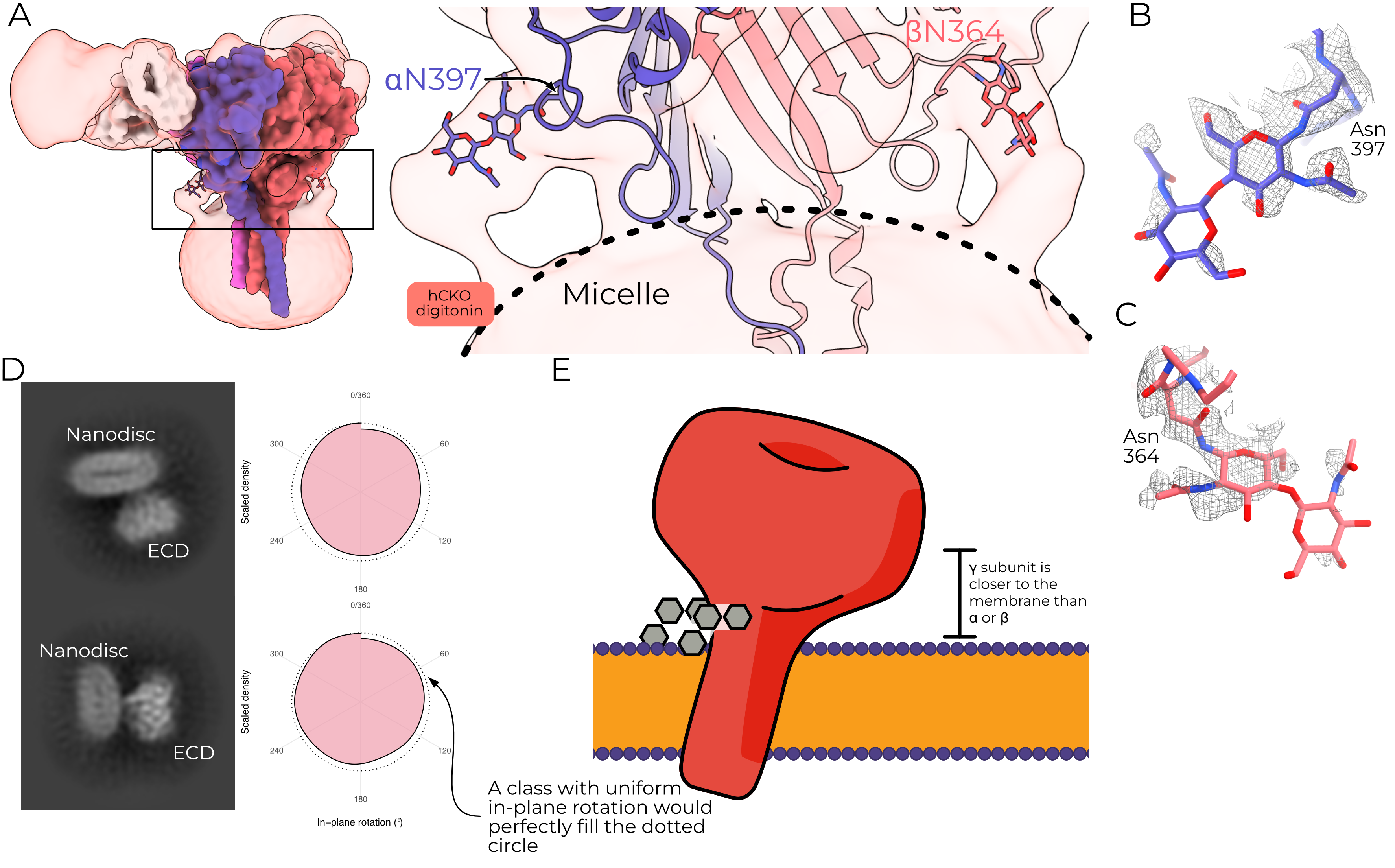
The micelle-proximal glycans are anchored to hαN397 and hβN364 and the equivalent mouse residues, mαN424 and mβN362 (Figure 4.10 A). The α site is conserved in mammalian ENaCs, while the β site is conserved in all surveyed organisms except lungfish. These glycans extend away from the ECD body and turn down toward the micelle or lipids and are clear in all ENaC maps solved to date. The γ subunit does not have an equivalent asparagine residue, and so does not have a micelle-proximal glycan.
I note that all ENaC maps produced to date also show a consistent tilt of the micelle or nanodisc such that the γ subunit is closer to the surface of the micelle than are the α or β subunits; this tilt does not appear to be an artifact of in-plane rotation (Figure 4.10 B). I propose therefore that this tilt may in part be caused by the presence of the micelle-proximal glycans, which are only missing from γENaC (Figure 4.10 C). Their presence in the nanodisc map may indicate that they perform a similar role in a native cell membrane context.
4.4 How does this change our understanding of the ECD?
4.4.1 A misfolded TMD may uncouple ECD motions
Regardless of the treatment applied, I cannot measure any change in the ECD. This stability stands in stark contrast to the highly dynamic ECD of the gating model widely assumed in the field and based on that of ASIC12,14. I observe trypsin cleavage of both the α and γ GRIP domains, but only reduced cryoEM map density for the γ domain. The maps are otherwise perfectly superimposable, with unchanged ECDs and TMDs. Loss of density for only the γ GRIP domain is surprising but not entirely unexpected — channels lacking the γ subunit or specific lysine residues in the γ ECD are not activated by trypsin3. It is therefore possible that while the α GRIP domain is cleaved, it is not directly involved in trypsin-mediated channel activation. Based on biochemical data, I speculate that the α GRIP domain is also cleaved by trypsin, but the GRIP domain is still clearly visible in our cryoEM maps. This could be the result of non-covalent interactions between the α GRIP domain and the α finger and thumb. In any case, a lack of any observable channel rearrangement upon removal of the γ inhibitory peptide (a state in which ENaC’s PO approaches 0.9) must lead us to a deeper consideration of ENaC gating15.
Although the best-studied mechanism of ENaC opening is GRIP proteolysis, multiple studies have found other stimuli which open the channel, including laminar shear stress and phospholipids (see Section 2.4 for more detail). ENaC PO is also sensitive to changes in both intracellular and extracellular pH16. It is thus possible that the cell possesses multiple mechanisms with which it can open ENaC and uses them in combination to tune Na+ permeability.
The possibility of a more complex pathway to open ENaC is supported by in vivo studies as well. The proportion of cleaved γENaC from mice lacking SGK1 is significantly smaller than that of WT mice, but cells from these kidneys have indistinguishable amiloride-sensitive current17. Similarly, ENaC missing the canonical furin cleavage site in the γ subunit is not cleaved when expressed in X. laevis oocytes, but currents do not differ between the mutant channels and WT18.
Indeed, in different tissues and in different studies the cleavage of either α- or γENaC seems to be of greater importance, hinting at an environmental effect on the relationship between GRIP cleavage and channel PO15,19,20. Based on this prior research and the maps I present here, I suspect that ENaC gating is not as simple as the proposed mechanism in which a single, coordinated protein maturation step moves the channel from a low- to high-PO state21. However, I cannot make firm conclusions about ENaC gating due to the unresolved question of the TMD.
As discussed in Chapter 3, the TMDs of all six models are disturbed and likely do not represent a physiological conformation. It is possible that the ECD requires some kind of feedback from the TMD, and that this coupling has been lost during purification and grid preparation.
Mutations in linkers between the TMD and ECD of other channels have dramatic effects on ECD function; for instance, the lurcher mutation in the GluR1 ECD/TMD linker dramatically increases ECD affinity for glutamate22. I suspect that the ENaC ECD requires a fully-assembled six-TM pore region to respond to GRIP domain excision, but such a conclusion would require deeper investigation. A TMD/ECD decoupling would also explain the lack of ECD shift with the introduction of the DEG mutation, which has been shown to hold the channel in an open state, and for which I see dramatic TMD restructuring but no change in the ECD23.
I speculate, based on these results, that the structures of both human and mouse ENaC are deeply dependent on some input from the membrane. We know that ENaC requires PIP2 to open, and is opened when bound by PIP3 regardless of cleavage state (Section 2.4.2.1). It would not be entirely surprising, then, if other membrane features inform ENaC’s folding state. It is possible that finding a more-stable ortholog, or one with a gating mechanism less dependent on membrane lipids, would improve the quality of the TMD, as our mouse maps have significantly better TMD density than our human maps.
4.4.2 A role for the β9-α4 linker in gating
In Malacoceros fuliginosus FaNaC, a histidine in the β9-α4 linker forms contacts with both TMs24. This interaction blocks channel opening until ligand is bound. In ENaC and Helix aspersa FaNaC the equivalent residue is a tyrosine, while in ASIC it is a tryptophan25. It is possible that any bulky or aromatic residue plays a similar ECD-lock role in this β-turn. In my models, only the β-subunit tyrosine is close enough to hydrogen bond with the TMs as I have modeled them, but this could be an artifact of the disordered TMD, or the interaction may be mediated by lipids.
I also note that the proline directly preceding this tyrosine in both the β and γ loops (α does not have a proline in this loop) is in the cis conformation. This is true in both human and mouse ENaC, in all treatment conditions. Our previous models missed this conformer, likely because of significantly poorer resolution. ASIC’s tryptophan in this position is also preceded by a cis-proline2. In light of the important role this loop plays in FaNaC gating, and the conserved cis-Pro/aromatic pair in ASIC and ENaC, further investigation of this loop may prove fruitful.
4.4.3 The acidic pocket’s uncertain role in sodium self-inhibition
Previous reports suggested that rearrangement of an acidic pocket in the α finger plays a key role in SSI5. Our K+ maps are solved in the presence of sodium levels far below those known to release SSI, but the acidic pocket does not rearrange. However, there are species differences in ENaC SSI, and the functional work describing this acidic pocket was all performed on mouse ENaC, while our high- and low-Na+ models are both of human ENaC4,5,8.
In human (but not rat) ENaC, pH titration changes the magnitude of SSI but, crucially, not the Ki of Na+; a surprising result if surface-accessible acidic residues form the SSI Na+-binding site. Moreover, ENaC cleavage essentially abrogates SSI, and residues in the upper finger which interact with the GRIP domain are implicated in SSI4,26,27. Despite this relationship, I also see no conformational shift of the acidic pocket upon trypsin treatment.
It may therefore be that the acidic pocket simply has a higher affinity for Na+ than K+ and therefore presents a higher barrier to ECD conformational shift when bound to Na+. This would effectively trap the channel in a closed state, rather than some third sodium self-inhibited state. Without observing the complete gating cycle of ENaC and the concomitant conformational changes it is impossible to make any firm conclusions about the precise mechanism of SSI. I therefore hope that our models will serve as a useful guide in future studies on this acidic pocket, as well as a search for other potential sources of SSI.
4.4.4 Glycans may further differentiate ENaC subunits
These maps represent the first structural study of ENaC’s extensive glycosylation. I must emphasize that these models all arise from particles which have only high-mannose glycosylation. However, glycosylation is required for ENaC function and trypsin digestion and our channels have the expected electrophysiological properties and respond to trypsin treatment in the expression system (Figure 4.11)9. I am therefore confident that the high-mannose glycans are sufficient for mechanistic inference.

Two distinct classes of glycan make observable interactions in our models: membrane-proximal and intersubunit glycans. I propose that the membrane-proximal glycans may help establish the micelle/nanodisc tilt observed in all ENaC maps to date, and that this tilt may have as-yet unknown functional implications.
I am not suggesting that these glycans alone are sufficient to define ENaC’s orientation in the membrane For example, in addition to missing a membrane-proximal glycan γENaC has two fewer residues between the palm and TM1 than do the α or β subunits28.
The β intersubunit glycan interacts with the top of the α thumb, which may serve to rigidify it. Indeed, knock-out of β glycans protects the α subunit from trypsin digestion9. Perhaps the β intersubunit glycan holds the α subunit in a pose which primes it for proteolytic cleavage. In contrast, the γ palm, not thumb, is rigidified by the other large intersubunit glycan. I note also that previous observation of channels cleaved by endogenous cellular machinery indicated that removal of the inhibitory peptide reduces the map quality for γ- but not αP3 and P46.
This alternate rigidification is interesting in the light of sodium self-inhibition. As yet, there is not a clear explanation for how mutating αH255 and γH233 (occupying the same structural position in their respective subunits) might have opposite effects on SSI (augmenting and eliminating it, respectively)29. Considering a rigid macro-domain comprising the uncleavable β subunit, γ palm, and α thumb (connected via glycans) and a flexible macro-domain comprising the α palm and finger and γ thumb, I see that γH233 belongs to the former, while αH255 belongs to the latter. Of course, more functional study is required to firmly establish this link, but I note that the SSI of channels lacking N-linked glycosylation sites is significantly reduced compared to WT9.