5 Conclusion
5.1 Context
ENaC is an essential regulator of sodium permeability in many tissues. It plays an important role in a number of biological processes and diseases, including hypertension and cystic fibrosis. Despite its importance, little is known about the exact molecular mechanism by which the channel opens and closes.
Unlike ENaC, a great deal of structure/function information is available for the close relative ASIC. It is understandable, then, that the field has long assumed the mechanisms of these two channels are similar. In the “ASIC-like” model, the GRIP domain forms a wedge which holds the finger and thumb apart. Proteolytic release of this wedge allows the finger and thumb to collapse. This collapse is transmitted through the palm to open a fenestration near the membrane, allowing ions to pour through the pore. Mutations in the ENaC palm have large effects on current in a pattern similar to that of ASIC1–3.
Proponents of this model (including our own lab) wave away differences between ASIC and ENaC currents. The most obvious is ASIC’s desensitization. ASIC closes less than a second after opening4. Contrast this with ENaC, which does not desensitize at all — an open channel will remain open essentially indefinitely5.
Desensitization in ASIC is in large part mediated by the four-residue β11-β12 linker2. This β-turn is quite well-conserved among ENaCs and ASIC1, with the important leucine and asparagine residues absolutely conserved, and its conformation in all models solved so far aligns well with ASIC’s open state6. There is one notable difference between the ASIC and ENaC linkers: an alanine is replaced with an acidic residue in all ENaC subunits and species. In currently-available structures this acidic residue does not form hydrogen bonds with other residues, but it may do so in the open conformation.
ENaC currents do diminish over time in the presence of high external sodium in a process called sodium self-inhibition (SSI). SSI is independent from Na+ currents and is relieved by proteolytic cleavage5,7. SSI seems to induce a large-scale rearrangement of the ECD, unlike ASIC desensitization8. Mutagenesis and cross-linking studies suggest that an acidic pocket comprising the finger and β-ball bind Na+ to close the channel9–11.
The precise relationship between ENaC and ASIC’s gating is, if anything, less clear as a result of the work presented in this document. With the highest resolution yet, we detect a clear lack of movement when large-scale rearrangements are expected. In the remaining space afforded to me, I will remind the reader what we’ve learned and wonder what it might mean for the field moving forward.
5.2 Contrition and Contemplation
5.2.1 What is actually happening with cleavage and self-inhibition?
Two things are true: when I remove the inhibitory peptide from the ECD, I see no large-scale rearrangement of the finger or thumb; and the transmembrane domain of this protein is almost certainly in some non-physiological state. The former finding runs contrary to years of cross linking, mutagenesis, and electrophysiology experiments; the latter should give us pause in throwing out the existing model.
Could it be, then, that ENaC’s ECD requires some kind of tension from or coupling to the membrane via the TMD? It’s hard for me to come up with a model in which the finger and thumb do move closer together in a physiological environment, but do not move together without tension from the membrane, but it is the simplest means by which we might resolve this conflict. It is also possible that gating involves a related, but different motion, as seen in FaNaC12.
Of course, this is all speculation. ASIC’s ECD and TMD are loosely coupled — proton binding and pore opening are related for less than a second. What could we do to probe the relationship between the finger, thumb, GRIP, and pore?
There exist a great number of ECD mutations known to modify ENaC’s PO regardless of open state. Some of these (e.g., the Liddle syndrome mutant αC479R) are in regions of the ECD for which our maps are consistently quite high-quality14. ECD-only maps of these mutations would provide valuable insight into the gating mechanism even without a visible TMD.
A better understanding of SSI would further augment our knowledge of protease-mediated channel opening. SSI is unique to ENaC, and so we have little to rely on for mechanistic insight. My finding that ions bind the acidic pocket in Na+ and K+ buffers is surprising, as is the lack of any significant rearrangement of the ECD. Cross linking studies indicate that ECD conformational shifts are required for SSI8. This leaves us in the same situation as with proteolytic cleavage: is some kind of mis-folding preventing the expected rearrangements, or do they simply not occur?
A number of SSI-abrogating or -augmenting mutations have been described in the literature. At this point, solving ECD structures with resolutions better than 3 Å is routine. Systematic investigation of map densities in channels with and without these mutations could identify promising SSI ion-binding sites which may, in turn, inform further mechanistic studies.
Native material is another obvious answer. Production of a map of high enough quality to analyze gating mechanisms necessitates (at the time of writing) that the material is purified. This brings us to the question of why the transmembrane domain is hard to see in the first place.
5.2.2 A new approach is needed for the transmembrane domain
A single model of the TMD would go a long way toward providing a believable model of ENaC’s gating cycle. As discussed in Chapter 3, I believe that solubilization with digitonin presents a significant obstacle to TMD resolution. FaNaC purified in n-dodecyl-β-D-maltoside (DDM) and exchanged into GDN and then 3:1 POPC:POPG nanodiscs yields a high-resolution TMD12. Our first maps of ENaC, also purified in DDM, provided the best TMD until the present work with mouse ENaC6.
However, the nanodiscs presented in this work have the worst TMD map of any presented. Moreover, my experience has demonstrated that ENaC is particularly difficult to get into nanodiscs in the first place when purified with DDM or digitonin. We know that the TMD is properly folded in the expression system, since it is the same system in which we perform electrophysiology. Purification with styrene-maleic acid may yield the first TMD map, as it yielded the first map of the re-entrant loop in ASIC15. Alternately, spiking in required lipids (especially PIPs) during solubilization may stabilize the TMD enough to get it into nanodiscs.
I do not think these minor changes alone will be sufficient. Rather, I believe future work should pursue homologues in parallel to efforts toward improved purification. In what I find difficult to believe is a coincidence, the authors of the FaNaC study published a phylogenetic analysis the year before they published their structure of one of the more distantly related FaNaC genes16. ENaCs exist as far back as jawless fishes; there is a wealth of orthologs to pick from17.
Despite their low macroscopic currents in exogenous expression systems, homomeric channels certainly seem to exist in the body (see Section 1.1, especially the tongue and blood vessels). A homomeric channel does away with the difficulties of using Fabs without introducing the concomitant difficulties of subunit assignment. Their small currents mean that the number of properly folded channels may be even smaller than for αβγ channels, but it’s probably worth checking.
5.2.3 Electrophysiology
Not everyone likes structure — that’s okay, there’s something here for you too. I have identified several glycans which, based on their extended visibility in my maps, may serve a structural role. We also know, as covered in Section 2.4.3, that glycans are necessary for sensation of laminar shear stress. Electrophysiological studies of ENaCs with single asparagine mutations have not been performed by any lab, to my knowledge. Using my maps as a guide, it may be fruitful to investigate the sensitivity of ENaCs lacking the most visible (presumably the most structurally constrained) glycans to LSS.
Additionally, the FaNaC paper raises an interesting question: is the bulky residue in the β9-α4 linker necessary for proper gating? Conservation of the surrounding residues and an adjacent cis-proline would suggest so. An investigation of this loop is interesting whether the channels can gate or not — the best kind of experiment.
5.3 Conjecture
What do I think should be done next, if I’m so smart?
5.3.1 We need a functional assay
Why doesn’t our channel seem to undergo structural changes when it’s sodium self-inhibited? Why can’t we see our TMD? Why doesn’t the GRIP domain release from the ECD when it’s cleaved? All of these questions and more are essentially unanswerable without knowing whether our channels are functional in the first place. All of the mysterious observations from my work are fully satisfied by the conjecture that we’re looking at misfolded garbage.
As such, my first priority moving forward would be the development and implementation of a functional assay. As I discussed in Section B.2, the SPA assay shows no sign of working any time soon, and isn’t even truly a functional assay.
The sodium flux assay is, on the other hand, already halfway to usefulness. Using a known-good ion channel (ASIC comes to mind, but any sodium-specific ion channel will do) would help push it to completion by isolating the “protein is good” variable from the “assay works” variable. With more confidence in the fact that our purified material has some bearing on what is present in the cells, we can move back to structural study.
5.3.2 Find a better construct for the TMD
As discussed in Section 3.4.2, the CKO and DEG mutations should both be abandoned. They never made sense in the first place, and they did not accomplish their stated goals of stabilizing the TMD and holding the TMD and ECD in open states, respectively. Returning to a wild-type-like construct will make our results more interpretable.
To find a satisfactory gene for further study, we should first perform thermo-FSEC assays on a wide range of ENaC orthologs. Thermal stability, like SPA, is not an assay for function. It is, however, high-throughput, and channels which are stable are more likely to remain functional once purified.
I would then take the most-stable 10% of these constructs and perform whatever functional assay we end up developing on all of them. Any gene with significant flux could be screened using the same cryoEM concentrations and grid parameters I’ve determined in this work. We thus know we’d be starting with the most functional, most stable material we can find, rather than picking genes at essentially random as was done previously. Any promising maps could have their purification and grid parameters refined while simultaneously performing proper electrophysiological studies to ensure selectivity and currents are within reason.
One potential objection to this method is that we do not know whether our symmetry-breaking Fabs will bind to all forms of ENaC. My response is two-fold. First, it is easy to determine this during the high-throughput thermo-FSEC experiments by adding a condition with Fabs and looking for a shift. Second, my Fab-less nanodisc dataset demonstrates that < 3 Å maps can be achieved without the use of Fabs.
Moreover, solving maps just to solve maps is pointless. Fabs bind tightly and predictably to human and mouse ENaC, and look where that got us.
5.3.3 There are other interesting regions of the channel
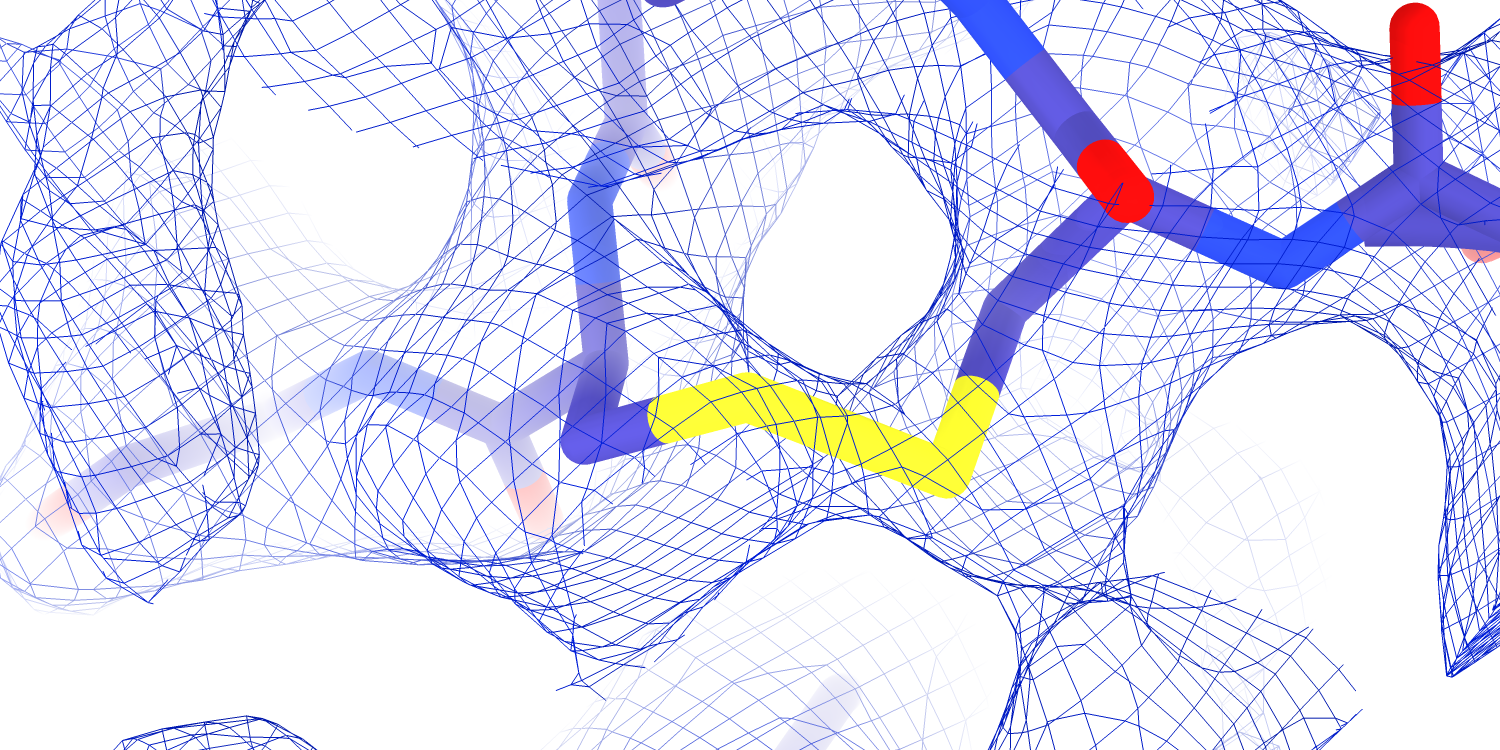
I think it was a mistake for me to focus exclusively on the TMD. Instead, there are exciting mutations in the ECD with described functional consequences that we already have map. For instance, mutation of a disulfide in the α subunit palm (αC479) results in Liddle syndrome, and we have high-quality map density for both halves of this disulfide (Figure 5.1).
5.4 Conclusion
After six years of work, I’m left with more questions than I had at the start. For decades, the ENaC field has been moving away from the gating model of “ASIC with an internal ligand”. Rather, ENaC PO is fine-tuned by a variety of internal and external signals. I believe that my findings here are consistent with that premise. I hope that this work is eventually a small piece of our understanding of this deceptively complicated channel.