1 ENaC Physiology and Regulation
As a guide to the importance of ENaC for human health, I will begin with a background in the physiological functions of the channel. Chapter 2 provides a deeper investigation of ENaC’s structure/function relationships. As a brief orientation, ENaC is a highly sodium-specific triheteromeric sodium channel expressed throughout the human body. The channel comprises one copy each of three paralogs: α-, β-, and γENaC. ENaC is thought to be activated by proteolytic removal of a portion of its extracellular domain and is potently inhibited by the small molecule amiloride and its derivatives.
I focus here on the systems in which we best understand ENaC’s role: the lung and kidney. I next move on to other systems which provide important information about ENaC but have a smaller body of research. I will finish the chapter with an overview of the molecular mechanisms of ENaC regulation at the expression level.
While this chapter may seem unnecessarily detailed for our purposes, I believe it is important to understand both ENaC’s role in several human diseases, the degree to which mouse models fail to recapitulate those diseases, and the shortcomings of viewing the channel only from the vantage of the kidney. This understanding is especially important when considering the limitations inherent to functional work performed in mouse tissue or on mouse proteins.
1.1 ENaC in the body
1.1.1 ENaC in the kidney
Blood pressure must be maintained within a narrow window of acceptable values. Too low, and vital organs do not receive sufficient oxygen and nutrients to function, but too high and blood vessels sustain damage. It is not surprising, then, that the human body has evolved several mechanisms for responding to changes in blood pressure, each with their own timescale ranging from milliseconds to weeks1. ENaC is the essential mechanism of the longest-term control, kidney excretion.
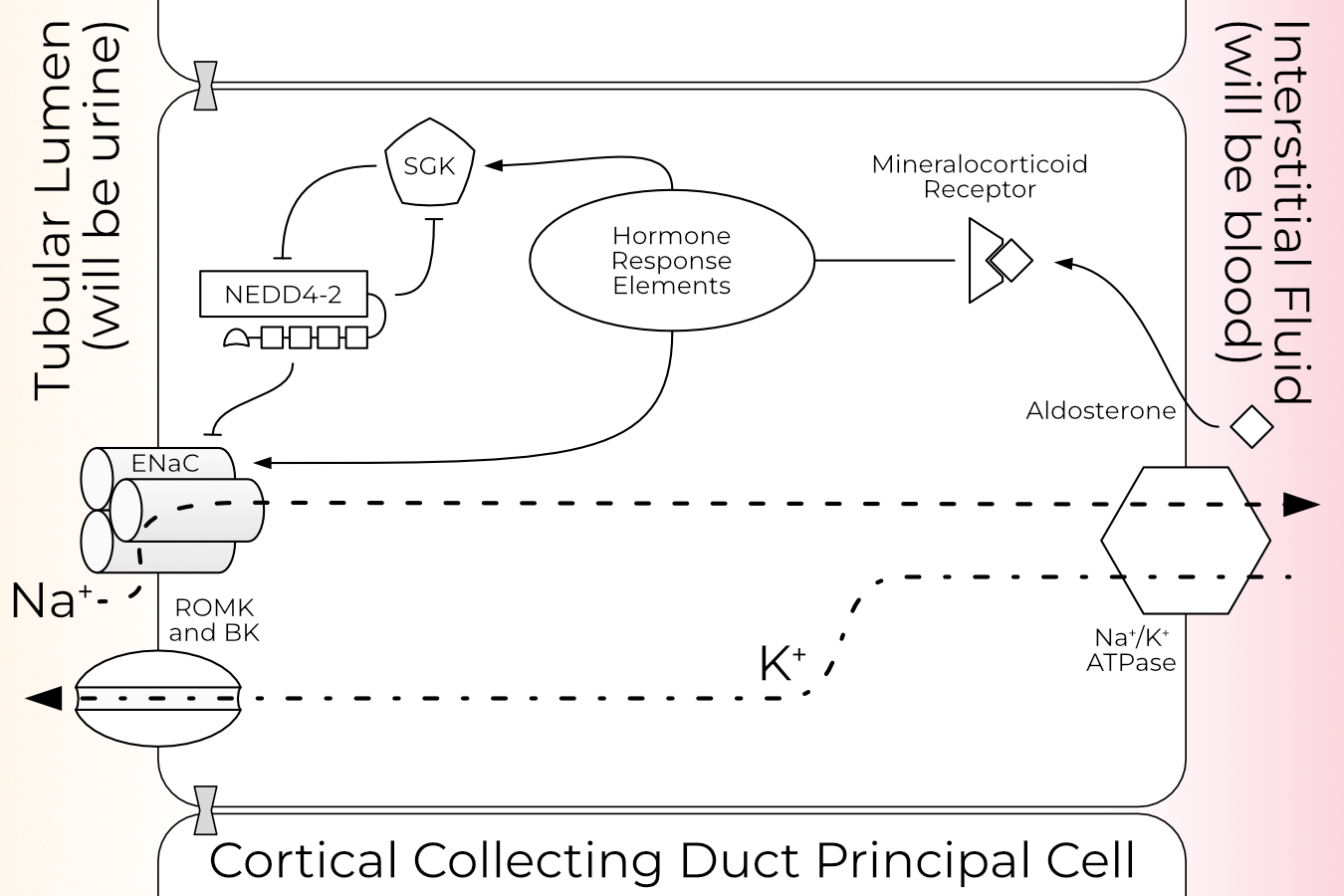
In the principal cells of the kidney, sodium travels down its concentration (into the cell) gradient across the apical membrane via ENaC (Figure 1.1). From there, it is transported across the basolateral membrane by the Na+/K+ ATPase. ENaC currents therefore induce retention of extra water to maintain the tightly controlled plasma sodium level. Thus, ENaC controls three essential functions of kidney filtration: first, the amount of sodium reabsorbed by the kidney; second, blood volume (and therefore pressure); and third, the amount of potassium passed from the plasma into the urine.
A variety of ENaC mutations have dramatic effects on patients’ blood pressure. One of the earliest described is Liddle syndrome2. Liddle syndrome (also called pseudoaldosteronism) results from an autosomal dominant gain-of-function mutation in ENaC. Hallmarks of the disease include severe hypertension, low potassium, high blood pH, low renin activity, and low aldosterone levels3. The disorder is thought to be rare, with only 72 families described as of 20184. However, after excluding patients with other clear causes (primary aldosteronism, kidney or heart diseases, and obstructive sleep apnea), approximately one in one hundred hypertensive patients had Liddle syndrome, indicating that the prevalence may be higher than is currently thought5,6.
All but one of the described cases involve mutation of the β or γ subunits4,6. A majority of these disrupt or remove entirely a proline-rich PY motif at the C-terminus of those channels7. This PY motif is the binding site for the E3 ubiquitin ligase Nedd4-2 (see Section 1.2.2). ENaC lacking the PY motif cannot be retrieved from the membrane, thus increasing sodium permeability by increasing N rather than the conductivity or PO of the channels8–10. The remaining minority of described Liddle syndrome mutations (including the sole ENaCα mutation) which do not affect PY-motif binding instead directly augment channel PO4. Liddle syndrome is typically treated with small molecules that block ENaC (amiloride or triamterene) and a low-salt diet.
Mouse models of Liddle syndrome complicate the situation somewhat. Mice with homozygous mutant Liddle βENaC fed a normal salt diet do not develop the characteristic high blood pressure, low potassium, and acidosis as humans do11. These phenotypes only develop with a high-salt diet. Moreover, these mice are still able to retrieve ENaC from the cell surface in the colon (unlike human Liddle patients)12. It is thus not clear that the mouse model of Liddle syndrome is fully applicable to the human disease.
Loss-of-function mutations also cause severe phenotypes. Type 1 pseudohypoaldosteronism (PHA1) was first described in a severely dehydrated infant who did not respond to aldosterone treatment13. There are two forms of PHA1: renal PHA1, which is milder and involves a mutation in the mineralocorticoid receptor; and systemic PHA1, which involves a mutation in a gene for ENaC α, β, or γ14,15. Patients with systemic PHA1 are unable to retain any salt, and so become severely dehydrated and have high potassium, low sodium, and increased acidity in their blood3. This makes the disease particularly deadly to newborns. Treatment typically includes life-long supplementation with sodium and the potassium elimination drug Kayexalate16.
Contrary to the pattern observed for Liddle Syndrome, most systemic PHA1 mutations occur in the gene encoding ENaCα16. The majority of described PHA1 mutations are nonsense mutations, although three missense mutations have been described15–17. One of these mutations occurs in the palm domain, one in the transmembrane domain, and one likely in the intracellular domain.
1.1.2 ENaC in the lung
Cystic fibrosis (CF) is a recessive genetic disorder which affects many organs, most prominently the lungs. Patients with CF have thick airway mucus they are unable to clear, causing chronic airway infections and lung damage. Patients with CF also lose salt but, unlike patients with PHA1, the causative mutation is in the CFTR gene rather than an ENaC gene.
CFTR encodes a cAMP-regulated chloride channel, CFTR18. Pathogenic CFTR mutations dramatically reduce chloride permeability, resulting in poor reabsorption of Cl- in the lungs19. Reduced chloride absorption increases the absorption of sodium to maintain electrostatic balance. The cells then absorb more water to maintain osmolarity. This, in turn, dehydrates and thickens the airway surface liquid (ASL), which damages the underlying tissue and prevents mucous clearance20–22.
The interplay between CFTR and ENaC in CF patients is complicated and not fully understood. Treatment of normal, but not CF, lung epithelia with amiloride augments Cl- transport23. Cells co-expressing CFTR and ENaC show smaller ENaC currents than do cells expressing only ENaC, but this effect depends on both CFTR’s ability to pass Cl- and ENaC’s intracellular termini24. Mice which overexpress βENaC in the airway show CF-like symptoms, including reduced airway liquid height and mucous clearance25. It would seem that the function of the two ion channels is linked both by electrochemical gradients and potentially other regulatory partners.
The relationship between ENaC-mediated Na+ absorption and CF or CF-like disease turns out not to be so neat. In mice which overexpress βENaC and CFTR, one would expect the CF-like symptoms from increased βENaC expression to be rescued by increased Cl- secretion. In actuality, said mice exhibit neither of these traits27. Additionally, Liddle syndrome patients have increased sodium absorption in all ENaC-expressing tissues but neither they nor Liddle syndrome mice develop CF-like lung disease28. In the opposite pattern of the mouse models, human patients with reduced βENaC function develop CF-like phenotypes of lung infection and elevated sweat chloride29. More confusingly, PHA1 patients (which have reduced ENaC expression in all tissues) produce excessive airway liquid as in CF-like disease, but this liquid does not become infected30.
There are many hypotheses as to how these contradictions may be resolved, reviewed well by Collawn and colleagues, but more work on the subject is desperately needed31. The important point for our purposes is that, as with Liddle syndrome, the mouse CF model does not recapitulate human disease well. Indeed, mice with CFTR mutations which are pathogenic in humans do not develop lung disease at all32.
1.1.3 ENaC in the tongue
As one might expect for a sodium-specific ion channel, ENaC is also implicated in taste. Although it may seem a less clinically essential tissue, ENaC function and expression in the tongue differs significantly from what is expected based on the better-studied kidney. It is therefore useful as a spotlight on how little we still know about the very basic stoichiometry and function of the ENaC subunits.
The organization of taste in the mammalian tongue is complex, but well-reviewed by Yarmolinsnky and colleagues33. Briefly, taste buds comprise 50–100 taste receptor cells (TRCs). Each TRC expresses receptors for and responds to just one of the five basic tastes: salty, sweet, sour, bitter, and umami34,35. These receptors are highly diverse, with closely related species having widely varying taste responses to the same molecule.
Salt sensation is further divided into an appetitive taste (e.g., a potato chip) and an aversive taste (sea water). Amiloride blocks rats’ ability to taste the difference between KCl and NaCl and reduces their voluntary NaCl (but not KCl) consumption37,38. Thus, the appetitive taste is taken to result from Na+ currents in amiloride-sensitive TRCs, while the aversive NaCl taste results from Cl- currents in amiloride-insensitive TRCs39. ENaC seems a prime candidate for the amiloride-sensitive Na+ current; indeed, all three subunits are detected in taste buds but not the surrounding epithelia40. Mice with αENaC specifically knocked-out in mature TRCs lost their preference for salt, and those TRCs no longer responded to low Na+ concentrations41. However, recent work has significantly complicated the role of αβγ ENaC in salt taste.
Investigation of ENaC expression patterns in mouse taste buds show that no taste bud cell expresses both the α and β subunits42. αENaC homomeric channels pass small (100 times smaller than αβγ) currents when expressed alone in oocytes43. Homomeric α channels may, therefore, carry the amiloride-sensitive salt taste. However, amiloride-sensitive salt perception depends on the mouse line, and the line used in the expression pattern study has a low amiloride sensitivity44. Thus the possibility remains that salt taste in mice is mediated by αβγENaC and is simply harder to detect with this experimental setup. Homomeric α channels may instead carry the amiloride-sensitive salt taste.
Perhaps more importantly, salt taste in humans is insensitive to amiloride45. Additionally, humans express δENaC (a fourth subunit which is a pseudogene in rats and mice) in the taste bud, rather than αENaC46. As with CF and Liddle syndrome, the mouse model of taste is a poor facsimile of the process in humans.
Despite this fact, salty taste illustrates an important unanswered question regarding ENaC: what is the full complement of physiologically relevant, functional ENaC channels? Canonical ENaCs are composed of one each of the β, γ, and either α or δ subunits. However, the combination of αENaC knock-out mice and ENaC expression patterns makes it likely that channels with only one or two ENaC subunit types, while rare, are physiologically relevant.
I note here that we cannot say these channels are homomeric αENaC channels. They may be assemblies of ENaC with other ENaC/DEG family members or as-yet unknown other proteins. Regardless of the ultimate identity of the amiloride-sensitive salt taste receptor, the patterns observed in taste are difficult to explain if the only physiologically relevant ENaC is the (α/δ)βγ triheteromer, which makes it an interesting and valuable system for further study.
1.1.4 ENaC in blood vessels
Myogenic vasoconstriction is an important component of blood pressure regulation in which the diameter of a blood vessel decreases in response to increased blood pressure. This response is essential for maintaining proper blood flow. ENaC is expressed in the vascular smooth muscle cells responsible for the constriction of blood vessels47,48. Treatment with amiloride or its derivatives reduces myogenic tone in a dose-dependent manner. This fact, combined with the knowledge that channels related to ENaC are mechanosensitive in worms, makes ENaC a likely candidate for the pressure sensor responsible for myogenic vasoconstriction49.
Similarly, systemic blood pressure is monitored by the central nervous system via baroreceptors. Baroreceptors keep mean arterial pressure at a steady level on a short time scale50. These neurons express β and γ, but not α, ENaC51. Treatment with amiloride or its derivatives blocks both the response of these neurons to physical stimulation and blood pressure regulation. It therefore seems likely that ENaC is used by baroreceptors to sense blood pressure, but further research is necessary to strictly define this role.
1.1.5 ENaC in the colon
ENaC plays an important role in sodium reabsorption in the colon. Colon expression of the β and γ subunits increases in response to aldosterone treatment, while α expression remains constant12,52. Patients with Crohn’s disease, a chronic inflammatory bowel disease, have reduced sodium reabsorption in the colon, which leads to diarrhea53,54. Similarly, patients with ulcerative colitis have reduced ENaC expression due to inflammatory cytokines, resulting in water loss and diarrhea55. To be clear, the colon relies on a wide array of systems to reabsorb ions and water. These results indicate that ENaC is one of the players in this process.
1.2 Regulation
It should come as no surprise that expression of ENaC is under tight control, given the number of important processes ENaC is implicated in. In this section I provide a general, by no means exhaustive review of proteins known to regulate ENaC at the expression level. A discussion of molecular mechanisms by which ENaC PO is modulated benefits from further background in ENaC structure and is therefore covered in Section 2.4.
This overview of ENaC regulation is necessarily brief and limited to the kidney (Figure 1.1), since that system is best understood. Fortunately, the patterns observed in the kidney largely hold in other tissues with the notable exception of aldosterone-induced expression.
1.2.1 Aldosterone
Aldosterone is the primary mineralocorticoid hormone and the main mechanism by which the body regulates plasma sodium levels. When the kidney is not receiving enough salt, it initiates a cascade to (among many other effects) increase activity of ENaC via the hormone aldosterone. ENaC is also regulated by other hormonal signals, including vasopressin and insulin, but a review of those mechanisms is outside the scope of this work. For the interested reader, Garty and Palmer’s review of the complete picture of ENaC regulation is quite thorough if a bit old58.
Aldosterone response is divided into two phases, aptly named the early and late phases. In the early phase, aldosterone increases ENaC PO in part via increased activity of serum and glucocorticoid-regulated kinase (SGK, Section 1.2.3)59,60. SGK’s effect has mainly been attributed to inhibition of Nedd4-2 (Section 1.2.2), while some labs also report a direct increase in ENaC PO61,62. Aldosterone also stimulates phosphatidylinositol 3-kinase (PI3K) and K-Ras, which promotes the physical interaction of ENaC and PI3K63,64. Phospholipids have both short- and long-term effects on ENaC currents and are discussed further in Section 2.4.2.1. The majority of aldosterone’s early-phase effect on sodium currents is due to increased PO of existing channels, rather than delivery of new channels to the cell surface65.
Aldosterone’s late phase increase of sodium permeability is mediated both by direct increase of ENaC gene expression and movement of ENaC from an internal pool to the apical membrane66–69. In some studies, aldosterone increases transcription of the β- and γ- (but not α-) ENaC genes12,66,70–72. In others, increases in αENaC transcription are also (or exclusively) observed69,73–77. The effect of aldosterone does seem to be consistent within tissue types: most studies demonstrating an increase in αENaC transcription are performed in the kidney, while aldosterone increases β and γ transcription in the colon. ENaC transcription in the lung is less dependent on circulating aldosterone levels than other tissues78.
1.2.2 Nedd4-2
Nedd4-2, also known as Nedd4L, is a HECT E3 ubiquitin ligase. E3 ligases perform the final step in the ubiquitination cascade: the transfer of an activated ubiquitin (Ub) from an E2 to the substrate. Nedd4-2 is the cognate E3 ligase for ENaC in humans, rats, and mice (but not Xenopus). Ubiquitination of ENaC by Nedd4-2 results in internalization and degradation of the channel, reducing the cell’s Na+ permeability. Study of ENaC surface dwell time is complicated by a reserve pool maintained by the cells to be cycled up to the membrane, but there is a consensus that ENaC is recycled quickly; surface half life estimates range from fifteen minutes to three hours, with the low end having more support79. Disruption of this mechanism is the most common cause of Liddle syndrome (Section 1.1.1), indicating the essential role Nedd4-2 plays in regulation of ENaC surface expression.
1.2.2.1 Nedd4-2 Structure
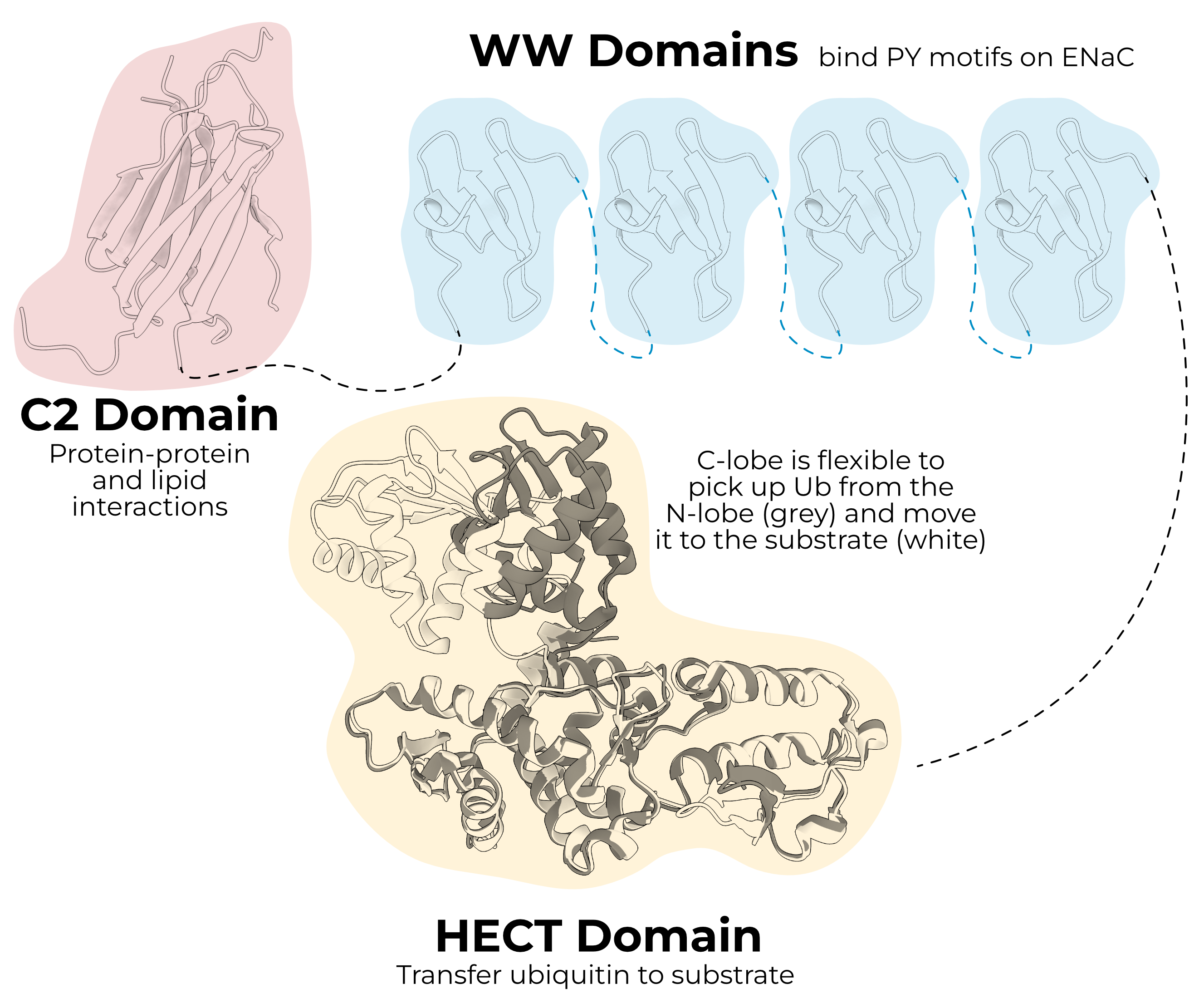
Like many members of the HECT ligase family, Nedd4-2 has an N-terminal C2 domain, three or four (depending on the species) WW domains, and a C-terminal HECT domain (Figure 1.2). As the astute reader has already guessed, humans also express a paralogous HECT E3 ligase, Nedd4, which does not regulate ENaC in vivo80,81. It is important to note that expressing either of these two ligases heterologously does reduce ENaC currents. This makes evaluation of the Nedd4-2/ENaC literature complicated, as conclusions drawn from heterologous systems may not hold in the organism.
In any case, the two proteins are 63% identical, and although there are differences in substrate affinity, the function of the domains in an abstract sense is quite similar. As such, I will present research on both proteins, preferring results from Nedd4-2 when possible, and highlighting points where the findings may not be fully generalizable.
The C2 domain is a Ca2+-dependent phospholipid and protein-binding domain. Increased intracellular Ca2+ induces translocation of Nedd4 from the cytosol to the membrane83. This apical targeting is the result of an interaction between the C2 domain and Annexin XIIIb84.
WW domains are protein-protein interaction domains comprising a three-stranded β-sheet. These small domains bind PY-motifs (PPXY), including those found in each ENaC subunit (PPAY, PPNY, PPKY in human α, β, and γ respectively). It is the removal of these domains that causes Liddle syndrome in truncation mutants85. Nedd4-2 cannot bind the Liddle mutants, and so the cell cannot pull the open channels from the surface, resulting in a gain of function. Interestingly, mice and rat Nedd4-2 have three WW domains, while human Nedd4-2 has four.
The HECT domain is the business end of the protein. Unlike RING ligases, HECT ligases form a thioester bond with Ub before transferring it to the substrate. The domain itself is split into the N-lobe, which is larger and interacts with the E2 enzyme, and the C-lobe, which contains the catalytic cysteine.
HECT ligases have several auto-inhibitory functions to prevent inappropriate ubiquitination. The C2 domain intramolecularly binds the HECT domain near the catalytic cysteine, blocking function87. The HECT domain also has an internal PY motif which is bound by its own WW domain88. Nedd4-2 remains bound this way until it encounters a substrate, at which point it binds and ubiquitinates the target. The enzyme then ubiquitinates itself, targeting it for degradation. Finally, the linker between WW domains 2 and 3 blocks function of the HECT domain in Nedd489.
1.2.2.2 Nedd4-2 and ENaC
Nedd4-2 is expressed throughout the body but is most abundant in the kidney and lung90. In a high-salt diet (i.e., low-aldosterone), the WW domains bind ENaC’s PY motifs and ubiquitinate the channels, causing them to be internalized and degraded61,91–93.
The functional importance of lipid binding by the C2 domain is unclear. In heterologous systems, both Nedd4 and Nedd4-2 inhibit ENaC currents with or without a C2 domain, and both rats and humans express splice variants of Nedd4-2 which is missing the C2 domain94–97. In fact, C2-less Nedd4 suppresses ENaC currents more than WT Nedd4 in heterologous systems94,98. This may indicate that the C2 domain is more important as a regulatory, rather than functional, domain. However, mice expressing Nedd4-2 without a C2 domain fail to regulate ENaC in response to a high-salt diet and develop hypertension, leaving open the possibility that the membrane-binding functionality is more important in vivo99.
Nedd4-2 relies on a subset of its WW domains to bind and regulate ENaC. In humans, only WW3 and WW4 of Nedd4-2 are required to reduce ENaC currents100. Human WW2 does bind ENaC weakly, while WW1 does not seem to interact with any ENaC subunit. Differential affinity among the WW domains is observed in other species as well94,101.
Once ENaC is bound, the HECT domain begins ubiquitinating N-terminal lysines on the channel93. Although lysines in each subunit are ubiquitinated, the γ subunit lysines are required for ubiquitination of α- and βENaC.
Several constitutively-expressed deubiquitinating enzymes (DUBs) oppose ENaC ubiquitination102–104. Interestingly, DUBs increase the relative amount of cleaved (i.e., active; see Section 2.4.1) ENaC at the cell surface. It is not known whether this effect is a direct result of DUB binding and action, or a simple follow-on effect of longer surface dwell time due to reduced recycling.
HECT domains are capable of serial ubiquitin addition, in which the C-terminus of ubiquitin is connected to the N-terminus or one of several lysines on the nascent chain105. Different chains determine the fate of the tagged protein; Nedd4-2 preferentially produces chains of ubiquitin connected by lysine 63 (K63 chains)106. K63 chains most often send substrates somewhere other than the proteasome for degradation or recycling. In the specific case of ENaC, some groups have reported lysosomal degradation, while others report a combination of proteasomal and lysosomal targeting92,93,108. Regardless of ubiquitinated ENaC’s ultimate destination, the interplay of Nedd4-2 and ENaC is critical to the overall sodium permeability of the cell.
1.2.3 SGK and 14-3-3
Serum- and glucocorticoid-induced kinase (SGK) is a serine/threonine kinase expressed in several tissues including heart, brain, lung, and kidney109. Transcription of SGK is directly induced by a multitude of signals, including aldosterone60,62,110.
Mice lacking SGK present a salt-wasting phenotype and high serum aldosterone, similar to the symptoms seen in PHA1 patients (Section 1.1.1)111. SGK increases ENaC surface expression and currents by disrupting the Nedd4-2/ENaC interaction by phosphorylating Nedd4-277,81,112. SGK is itself, in turn, ubiquitinated by Nedd4-2114. This establishes a tight negative feedback loop which imparts a sharper temporal dependence of SGK expression on aldosterone.
SGK also directly interacts with and phosphorylates αENaC115,116. This association increases ENaC activity even in channels lacking the C-terminal PY motif117.
Phosphorylated Nedd4-2 is bound and inhibited by 14-3-3, which is a protein that binds other phosphorylated regulatory proteins118,119. Humans express seven isoforms of 14-3-3; at least the β, ε, and η isoforms are known to dimerize and regulate Nedd4-2120,121. 14-3-3 binds with a high affinity to Nedd4-2 phosphorylated at serines 342 and 448.
14-3-3 binding rearranges and reduces the solvent accessibility of WW domains 3 and 4 (which are essential for ENaC binding) and the catalytic cysteine121,122. It is likely, then, that 14-3-3 regulates Nedd4-2 both by blocking its catalytic activity and sterically preventing Nedd4-2/ENaC.
1.3 Conclusion
ENaC is a tightly regulated ion channel implicated in a vast array of biological functions, from blood pressure control to the pleasurable taste of salty food. I have provided here a brief overview of the tissues in which ENaC expression is responsive to aldosterone (the body’s main salt regulatory hormone) as well as the proteins involved in coordinating its expression.
I hope I have also highlighted the great amount we do not know about the channel. ENaC’s function and regulation in the kidney is well-studied, but the trends observed in that tissue do not translate well to the lung (in which Liddle syndrome patients do not develop disease) or tongue (in which no heterotrimeric channels seem to exist). Additionally, the transcriptional and translational responses to aldosterone differ in different tissues.
Regardless, a deeper understanding of ENaC is necessary for a deeper understanding of the disorders and diseases covered in this chapter. To that end, I next present some background on what we know about ENaC currents and what we can infer about its molecular mechanism of gating based on related channels.